Quick Start
Quick Start to use the web interface
Navigate to https://rgetoolkit.com/
Download the provided Sample VCF file
Default settings:
Target Genome: Homo sapiens (GRCh38/hg38)
PAM Type: SpCas9 from Streptococcus pyogenes: 5’-NRG-3’
Query Sequence: CAGCAACTCCAGGGGGCCGC
Mismatches: 3
Click Submit to process the sample file.
For custom analysis, upload your phased single-sample VCF file.
For faster execution, upload a VCF file containing a few chromosomes, like chr1 and chr2, by filtering them using the command:
bcftools view -r chr6,chr10 NA12878.vcf.gz -o Output.vcf.gz
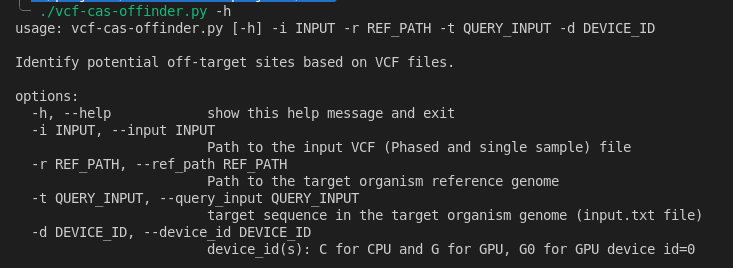
Quick Start to use the CLI
Create a conda environment
conda create -n varcasoffinder
Activate the conda environment:
conda activate varcasoffinder
Install all dependencies
pip install -r requirements.txt
Then for help, run:
./vcf-cas-offinder.py -h